IDTテクニカルレポート vol.2

>> PDF版はこちら
はじめに
ゲノム編集技術は、様々な生物種において自由な遺伝子操作を可能にし、基礎から応用まで多岐にわたるメディカル、ライフサイエンス研究に影響をもたらしている。マウス・ラットを含む様々な実験動物においても、従来のES細胞を用いた遺伝子改変に比べ、より簡単、効率的かつ短期間で遺伝子改変動物を作製できるようになった。特に2012 年に報告されたCRISPR/Cas9システムは、変異導入効率の高さに加えて低コストかつ簡単に利用できることから、現在ではゲノム編集ツールの中心となっている。
CRISPR/Cas9を用いた遺伝子改変マウス・ラットの作製法は極めてシンプルである。受精卵にCas9 mRNAおよびgRNAをマイクロインジェクションにより導入し移植をして得られた産子から変異個体を選抜する。標的領域と相同配列を持つドナーDNAを同時に導入することで、ノックイン個体も作製できる。
ここでは、筆者らが新たに開発した効率的なプラスミドノックイン法を紹介するとともに、gBlocks®、Ultramer®を用いたクローニングを必要としない簡便な遺伝子改変動物の作製方法について解説する。
CRISPR/Cas9を用いた遺伝子改変マウス・ラットの作製法は極めてシンプルである。受精卵にCas9 mRNAおよびgRNAをマイクロインジェクションにより導入し移植をして得られた産子から変異個体を選抜する。標的領域と相同配列を持つドナーDNAを同時に導入することで、ノックイン個体も作製できる。
ここでは、筆者らが新たに開発した効率的なプラスミドノックイン法を紹介するとともに、gBlocks®、Ultramer®を用いたクローニングを必要としない簡便な遺伝子改変動物の作製方法について解説する。
gBlocks®によるクローニングフリーgRNA作製
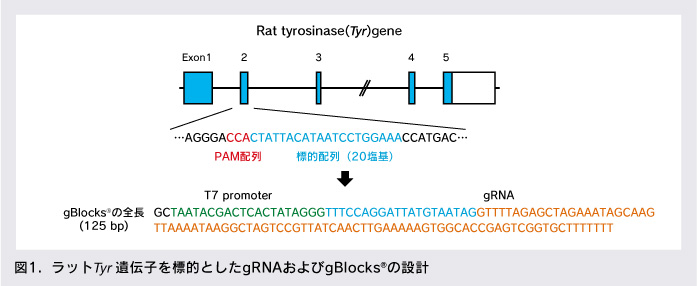
はじめに標的遺伝子、標的領域のDNA配列情報をEnsemblやUCSC等から入手し、それらを認識するgRNAを設計する。筆者らは、主にCRISPR Design Tool(http://www.genome-
engineering.org/crispr)を用いてgRNAを設計している。現在では様々なgRNA設計ツールが充実しており、複数のツールを利用することで、オフターゲットへの影響を十分に考慮した標的配列を決定できる。
その後、T7プロモーターおよび標的を認識するgRNAをコードした全長125塩基の二本鎖DNAをIDT社のgBlocks®で注文する(図1)。この際、合成できない配列を含む場合はその配列を示してくれるため、その場で標的配列を設計し直すことができる。gBlocks®は1週間程度で納品され、クローニングの必要がなく、すぐにin vitro転写に利用できる。筆者らは届いたgBlocks®を10 µLのNuclease Free waterで希釈後、4回に分けて転写の鋳型に利用しているが、インジェクションに十分量のgRNAを合成できている。一方Cas9についても、T7プロモーター下Cas9発現プラスミドを用いてCas9 mRNAを合成する。筆者らはaddgene社より入手したCas9発現プラスミドを改良して高効率に変異を導入できるものを使用している[5]。
ノックイン動物の作製では、Cas9 mRNAおよびgRNAに加えて、標的領域と相同配列を持つドナーDNAを同時に導入する。詳細は後述するが、ドナーDNAには、一本鎖オリゴヌクレオチド(ssDNA)もしくはDNAプラスミドが用いられる。ssDNAを用いる場合は、gBlocks®と同様、設計したものをIDT社のUltramer®で発注するだけで、納品されたものを希釈後、すぐに使用できる。
インジェクション溶液は、状況に応じて、50-100 μg/mL Cas9 mRNA、25-50 μg/mL gRNA、25-50 μg/mL ssDNA、3-5 μg/mLプラスミドDNAになるようRNase free waterで調製する。マウスもしくはラット受精卵に対し、混合溶液を顕微注入する。一晩培養後、2細胞期胚を偽妊娠メスに卵管移植し、3週間程度で、産子を得ることができる。
3週齢程度の得られた産子から採血、あるいは尾切り等により組織を採取し、DNAを抽出する。このDNAを用いて標的領域のPCRを行い、電気泳動にてDNA増幅を確認する。さらに、PCR産物のダイレクトシークエンス解析を行い、遺伝子改変個体(ファウンダー)を選抜する。モザイクやヘテロに変異が入り、波形が重なる場合、PCR産物をTAクローニングし、複数の配列を解読する。
engineering.org/crispr)を用いてgRNAを設計している。現在では様々なgRNA設計ツールが充実しており、複数のツールを利用することで、オフターゲットへの影響を十分に考慮した標的配列を決定できる。
その後、T7プロモーターおよび標的を認識するgRNAをコードした全長125塩基の二本鎖DNAをIDT社のgBlocks®で注文する(図1)。この際、合成できない配列を含む場合はその配列を示してくれるため、その場で標的配列を設計し直すことができる。gBlocks®は1週間程度で納品され、クローニングの必要がなく、すぐにin vitro転写に利用できる。筆者らは届いたgBlocks®を10 µLのNuclease Free waterで希釈後、4回に分けて転写の鋳型に利用しているが、インジェクションに十分量のgRNAを合成できている。一方Cas9についても、T7プロモーター下Cas9発現プラスミドを用いてCas9 mRNAを合成する。筆者らはaddgene社より入手したCas9発現プラスミドを改良して高効率に変異を導入できるものを使用している[5]。
ノックイン動物の作製では、Cas9 mRNAおよびgRNAに加えて、標的領域と相同配列を持つドナーDNAを同時に導入する。詳細は後述するが、ドナーDNAには、一本鎖オリゴヌクレオチド(ssDNA)もしくはDNAプラスミドが用いられる。ssDNAを用いる場合は、gBlocks®と同様、設計したものをIDT社のUltramer®で発注するだけで、納品されたものを希釈後、すぐに使用できる。
インジェクション溶液は、状況に応じて、50-100 μg/mL Cas9 mRNA、25-50 μg/mL gRNA、25-50 μg/mL ssDNA、3-5 μg/mLプラスミドDNAになるようRNase free waterで調製する。マウスもしくはラット受精卵に対し、混合溶液を顕微注入する。一晩培養後、2細胞期胚を偽妊娠メスに卵管移植し、3週間程度で、産子を得ることができる。
3週齢程度の得られた産子から採血、あるいは尾切り等により組織を採取し、DNAを抽出する。このDNAを用いて標的領域のPCRを行い、電気泳動にてDNA増幅を確認する。さらに、PCR産物のダイレクトシークエンス解析を行い、遺伝子改変個体(ファウンダー)を選抜する。モザイクやヘテロに変異が入り、波形が重なる場合、PCR産物をTAクローニングし、複数の配列を解読する。
一本鎖オリゴヌクレオチドUltramer®による効率的ノックイン
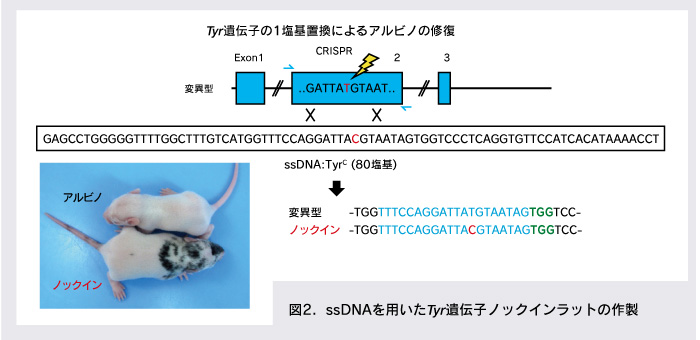
Tyr遺伝子エクソン2のSNP(赤色)を置換するために、80塩基のssDNAを設計した。ssDNAはSNPから上流39塩基、下流40塩基にそれぞれホモロジーアームを持つ。マイクロインジェクションの結果、ノックイン個体が得られ、毛色の発現がアルビノから修復した。青色:gRNAの認識配列。緑色:PAM配列。
ドナーDNAとして用いるssDNAの配列には、導入、改変したい配列に加えて、前後に相同配列を設計する必要がある。相同配列の最適な長さについては議論の余地があるが、筆者らは通常ノックイン領域の前後各40塩基の相同配列を設計、注文している。ssDNAは逆相カラム精製程度の純度であれば、ドナーDNAとして利用できる実績があるが、IDT社の高純度ssDNAであるUltramer®を用いた場合、脱塩グレードでも効率よくノックイン個体を作製できている。
例えば、アルビノラットであるF344系統は、毛色遺伝子であるTyr遺伝子に一塩基変異を有している。筆者らはこの変異を修復するため、野生型SNPおよび両側各40塩基の相同配列を持つssDNAを設計した。またgRNAの標的配列を、アルビノ型SNP上に設計しておくことで、ノックイン導入後に再度二本鎖切断を引き起こすことを防いだ。これらgRNA、ssDNAをCas9 mRNAと共に受精卵に顕微注入することで、一塩基置換されたTyr遺伝子ノックインラットが作製でき、実際に表現型も修復できた(図2)。この他に、ssDNAを用いて、Asip遺伝子の19塩基挿入、Kit遺伝子のレトロトランスポゾン除去といった複数のノックインラットを作製することに成功している[1]。
ssDNAは後述のプラスミドDNAを用いたノックインに比べてクローニングの必要がなく調製が簡単であるうえ、ノックイン効率も高い。そのため短い配列の挿入・置換には有用である一方、長鎖のssDNAは合成が難しく、Ultramer®でも最大200塩基である。今後、ssDNAの合成技術の発展に伴い、カセット遺伝子の挿入やGFPによる遺伝子タグ化など長鎖のssDNAを用いたノックインも可能になるだろう。
例えば、アルビノラットであるF344系統は、毛色遺伝子であるTyr遺伝子に一塩基変異を有している。筆者らはこの変異を修復するため、野生型SNPおよび両側各40塩基の相同配列を持つssDNAを設計した。またgRNAの標的配列を、アルビノ型SNP上に設計しておくことで、ノックイン導入後に再度二本鎖切断を引き起こすことを防いだ。これらgRNA、ssDNAをCas9 mRNAと共に受精卵に顕微注入することで、一塩基置換されたTyr遺伝子ノックインラットが作製でき、実際に表現型も修復できた(図2)。この他に、ssDNAを用いて、Asip遺伝子の19塩基挿入、Kit遺伝子のレトロトランスポゾン除去といった複数のノックインラットを作製することに成功している[1]。
ssDNAは後述のプラスミドDNAを用いたノックインに比べてクローニングの必要がなく調製が簡単であるうえ、ノックイン効率も高い。そのため短い配列の挿入・置換には有用である一方、長鎖のssDNAは合成が難しく、Ultramer®でも最大200塩基である。今後、ssDNAの合成技術の発展に伴い、カセット遺伝子の挿入やGFPによる遺伝子タグ化など長鎖のssDNAを用いたノックインも可能になるだろう。
ホモロジーアームフリーのプラスミドDNAノックイン(2H2OP法)
これまでにCRISPR/Cas9を用いたプラスミドノックインマウス、ラットは複数報告されている[2]-[4]。しかし、その効率は研究室間でばらつきがあり、ノックインしたい配列に加えて、約500 bp?数kbのホモロジーアームをプラスミドDNAに導入する必要がある。筆者らは、こうした問題を克服するため、ホモロジーアームを持たないプラスミドDNAの効率的なノックイン法を開発した。
この方法では、Cas9 mRNAに加え、ゲノムDNAおよびプラスミドDNAをそれぞれ認識する2種類のgRNA、2種類のssDNA、プラスミドDNAを一緒に受精卵に導入する(図3A)。その際、CRISPR/Cas9が「はさみ」としてゲノム上とプラスミド上の標的配列を切断し、二本のssDNAが「のり」としてゲノムとプラスミドを上流と下流をそれぞれ結合修復することで、プラスミドDNAを特定のゲノム上に正確かつ効率的にノックインできる。筆者らはこの、2つのgRNAでDNAを切断し、2つのオリゴDNAでプラスミドをノックインする方法を2ヒット2オリゴ法(2H2OP法)と呼んでいる。実際に、2H2OP法を用いて導入遺伝子を安定発現するRosa26遺伝子領域にCAG-GFPプラスミドを導入したノックインラットを作製することを試みた。その結果、17.6 %の効率でCAG-GFPプラスミドをラットRosa26遺伝子内にノックインすることができた[5](図3B)。得られたノックインラットは1コピーだけの導入にも関わらず非常に強いGFP発現を示しており、子孫へも安定的に伝達されている。マウスでも同様の実験を行い、GFPノックインマウスの作製に成功している。
2H2OP法は、既存のプラスミドにホモロジーアームを付加することなくそのまま利用することできるため、煩雑なクローニング等の作業が必要ない。すなわち、遺伝子改変動物作製の計画を立ててから、パソコン上で設計、発注後、届いたものを転写し、インジェクションを行うだけであり、非常に迅速かつ効率的に遺伝子改変動物を作製できる。また、これまでのES細胞等による遺伝子改変技術では困難であった200 Kb以上の長いBACプラスミドを用いたノックインにも成功している[5]。さらに、標的とするゲノム配列の上流と下流2箇所、プラスミド1箇所の合計3箇所を切断する3H2OP法に応用も可能で、長鎖の遺伝子クラスタをプラスミドに置換するなど、応用性は極めて高い。一方で、2H2OP法はゲノムとプラスミドの結合部位に変異が入りやすく、想定の配列通りにノックインする正確性には改善の余地がある。今後、こうした正確性や効率性を改良することで、汎用性の極めて高いノックイン作製法になると期待している。
この方法では、Cas9 mRNAに加え、ゲノムDNAおよびプラスミドDNAをそれぞれ認識する2種類のgRNA、2種類のssDNA、プラスミドDNAを一緒に受精卵に導入する(図3A)。その際、CRISPR/Cas9が「はさみ」としてゲノム上とプラスミド上の標的配列を切断し、二本のssDNAが「のり」としてゲノムとプラスミドを上流と下流をそれぞれ結合修復することで、プラスミドDNAを特定のゲノム上に正確かつ効率的にノックインできる。筆者らはこの、2つのgRNAでDNAを切断し、2つのオリゴDNAでプラスミドをノックインする方法を2ヒット2オリゴ法(2H2OP法)と呼んでいる。実際に、2H2OP法を用いて導入遺伝子を安定発現するRosa26遺伝子領域にCAG-GFPプラスミドを導入したノックインラットを作製することを試みた。その結果、17.6 %の効率でCAG-GFPプラスミドをラットRosa26遺伝子内にノックインすることができた[5](図3B)。得られたノックインラットは1コピーだけの導入にも関わらず非常に強いGFP発現を示しており、子孫へも安定的に伝達されている。マウスでも同様の実験を行い、GFPノックインマウスの作製に成功している。
2H2OP法は、既存のプラスミドにホモロジーアームを付加することなくそのまま利用することできるため、煩雑なクローニング等の作業が必要ない。すなわち、遺伝子改変動物作製の計画を立ててから、パソコン上で設計、発注後、届いたものを転写し、インジェクションを行うだけであり、非常に迅速かつ効率的に遺伝子改変動物を作製できる。また、これまでのES細胞等による遺伝子改変技術では困難であった200 Kb以上の長いBACプラスミドを用いたノックインにも成功している[5]。さらに、標的とするゲノム配列の上流と下流2箇所、プラスミド1箇所の合計3箇所を切断する3H2OP法に応用も可能で、長鎖の遺伝子クラスタをプラスミドに置換するなど、応用性は極めて高い。一方で、2H2OP法はゲノムとプラスミドの結合部位に変異が入りやすく、想定の配列通りにノックインする正確性には改善の余地がある。今後、こうした正確性や効率性を改良することで、汎用性の極めて高いノックイン作製法になると期待している。
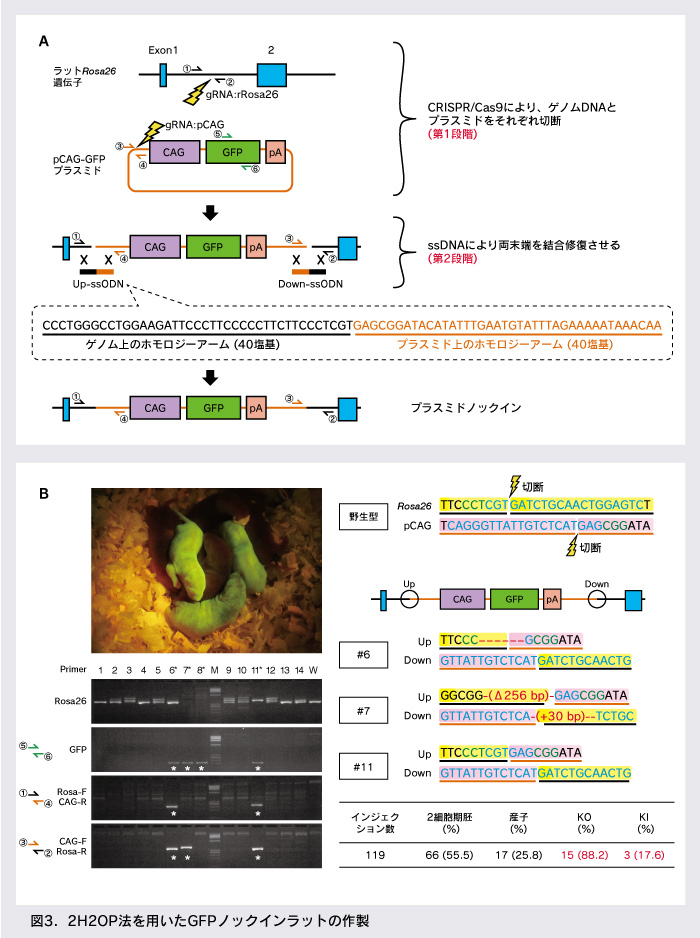
A. 2ヒット2オリゴ法(2H2OP法)の概要。ゲノム上およびプラスミド上にそれぞれgRNAを設計することで、ゲノムおよびプラスミドの切断末端が生じる。それぞれの末端部分を、ホモロジーアームを持つssDNAにより結合修復することで、プラスミドが標的部位へ導入されたノックインを得る。
B. 得られたGFPノックインラットとその遺伝子配列。#6、7、8、11のGFP陽性の4個体のうち、#11はssDNAの設計通りの配列でCAG-GFPプラスミドがRosa26領域にノックインされている。#6は上流、#7は上流、下流ともに欠失や挿入が見られるものの、CAG-GFPプラスミドがRosa26領域にノックインされている。
#8は標的領域部位にプラスミドの挿入が見られず、他の部位にランダム挿入されたと考えられる。
まとめ
2本鎖DNAであるgBlocks®を用いることでgRNA発現プラスミドのクローニング作業が必要なく、任意のgRNAを合成できる。同時にUltramer®で一本鎖オリゴDNAを設計・注文することで、ドナーDNAとして、もしくは今回紹介した2H2OP法のプラスミド挿入用としてすぐに利用できる。このようにCRISPR/Cas9を用いた正確かつ効率的なノックイン技術にgBlocks®やUltramer®を組み合わせることで、より簡便にノックイン動物を作製でき、遺伝子の生理的機能をより詳細に解析するための強力なツールとなるだろう。
参考文献
[1] K. Yoshimi et al. : Nat. Commun., 5:4240, 2014
[2] H. Yang et al. : Cell, 154 : 1370–1379, 2013
[3] Y. Ma et al. : FEBS J. 281(17) : 3779-3790, 2014
[4] T. Aida et al. : Genome Biol., 16(1) : 87, 2015
[5] K. Yoshimiet al. : Nature Commun.,7:10431, 2016
[1] K. Yoshimi et al. : Nat. Commun., 5:4240, 2014
[2] H. Yang et al. : Cell, 154 : 1370–1379, 2013
[3] Y. Ma et al. : FEBS J. 281(17) : 3779-3790, 2014
[4] T. Aida et al. : Genome Biol., 16(1) : 87, 2015
[5] K. Yoshimiet al. : Nature Commun.,7:10431, 2016
一本鎖オリゴヌクレオチド Ultramer®について
Ultramer®とは、200塩基まで合成できる一本鎖DNAオリゴヌクレオチドです。弊社の通常のDNA合成でも、業界標準よりも良い品質であると自信を持っておりますが、Ultramer®はさらに良い品質が期待出来ます。
>> 詳細はこちら
>> 詳細はこちら
人工遺伝子合成受託サービスgBlocks® Gene Fragmentsについて
直鎖上2本鎖DNA合成gBlocks® Gene Fragmentsはプラスミドに入っていない二本鎖DNAを合成するサービスです。安価で、1週間で納品できる事も多く、大変好評です。本レポートやテクニカルレポートvol.1の様に、CRISPR/Cas9システムにも多く利用されて参りました。
>> 詳細はこちら
2015年10月にさらにCRISPR/Casに特化したAlt-R™ CRISPR-Cas9 Systemの販売も開始しました。Alt-R™ CRISPR-Cas9 Systemを用いれば、さらに簡単に、効率良くゲノム編集を行えます。
>> 詳細はこちら
>> 詳細はこちら
2015年10月にさらにCRISPR/Casに特化したAlt-R™ CRISPR-Cas9 Systemの販売も開始しました。Alt-R™ CRISPR-Cas9 Systemを用いれば、さらに簡単に、効率良くゲノム編集を行えます。
>> 詳細はこちら