アプリケーションノート - HDR実験の最適化
1本鎖DNA(Ultramer®オリゴヌクレオチド)を用いる場合
真核細胞におけるゲノムの安定性には、効率的なDNA修復の機序が欠かせません。 二本鎖DNA切断を修復するには、二種類の経路が存在します。
比較的エラーを起こしやすい比相同的末端接合(NHEJ)経路と、遺伝情報を関連配列から正確にコピーしてDSBをエラーなく修復する、相同組換え修復(HDR)経路です。
目的遺伝子の機能欠損実験のために、フレームシフトまたは変異を標的遺伝子のオープンリーディングフレームへNHEJにより導入することによって、遺伝子機能を欠失させます。これは遺伝子「ノックアウト」と呼ばれるプロセスで、1ページを書籍から破り取ってしまうことにも似ています。
あるいは、選択可能なマーカー、蛍光タグ、または他の機能的単位などの外来性配列を、内在性のHDR経路によりシームレスに挿入し(「ノックインし」)ます。これは、一部では「真の」ゲノム編集と呼ばれています。
NHEJに基づくノックアウトは、研究ツールとして非常に十分に解明されており、比較的効率のよい手法ではありますが、HDRによる外来性DNAのノックインは、特に哺乳類の細胞において、課題が残っています[2–4]。
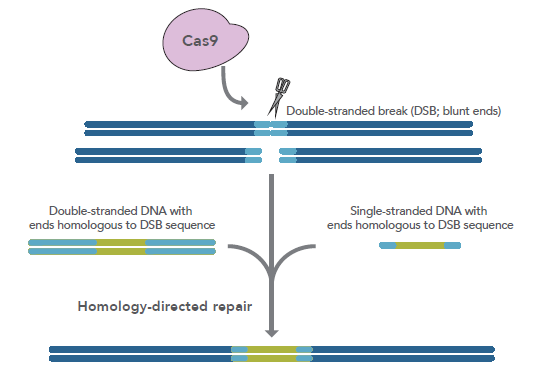
図1. 特定の遺伝子サイトにおける外来性ドナー配列のHDRによる挿入
Cas9(ピンク)は、gRNAとの複合体によって特定のターゲットを切断するように指定されている。
目的のインサート(緑)とホモロジーアーム(薄い青)サイトを含む、二本鎖または一本鎖のいずれかのドナーテンプレートとともにCas9:RNA複合体(RNP - リボヌクレオプロテイン)を導入すると、このドナーテンプレートは、HDR経路を介して目的のゲノム上に取り込まれる。(用語については下記を参照して下さい。)
十分な長さのホモロジーアームを含むこのドナーDNAは、図1に示されるように、Cas9リボヌクレオプロテイン(RNP)複合体(Cas9エンドヌクレアーゼとgRNAによって形成)と同時に導入されなければなりません[5]。
これらの情報は、エピトープタグなどの短い配列の挿入や、点変異の置換および修正に役立ちます。

用語解説
crRNAは、20塩基のプロトスペーサー配列(認識配列)と、tracrRNAに対して相補的な配列を有している。tracrRNAは、crRNAの相補配列とハイブリダイズし、複合体を形成する。crRNA:tracrRNA 複合体は、Cas9タンパク質とさらに複合体(RNP-リボヌクレオプロテイン)を形成する。このRNPの活性により、標的ゲノム内の特定の部位でDSB(二本鎖切断)を引き起こす。これら2種類のRNAは化学的に合成することができ、ゲノム編集に用いることができる。IDTの科学者達は、これら2種のRNAの長さおよび組成を改良し、ゲノム編集効率の最適化を図った。
ゲノム内でDSB(二本鎖切断)が起こった際のDNA修復メカニズム。相同組換えを介して、ドナーDNAをゲノムへ組み込む働きをもつ。この細胞内メカニズムを利用して、CRISPR-Cas9によって生じた特定サイトへのDSBに対し、目的の配列を挿入させる。
ゲノムDNAに導入したいインサート配列と、DSB周辺の相補的なホモロジーアームを両端にもつ1本鎖もしくは2本鎖DNA(図1)。ゲノムに対する点変異や、タグの付加、他の機能の挿入には、ドナーテンプレートが必要となる。
ゲノムDNAにおけるDSB修復機構で進化上保存されている。CRISPR-Cas9でおこるゲノム編集において、もっとも頻度の高い修復機構である。NHEJは、HDRとは対照的に修復に相同配列を必要とせず、DSB後のライゲーション時には、両端の数塩基の相同性を用いて修復を行っています。 NHEJは、エラーを起こしやすい修復経路であり、遺伝情報の精確な導入が望まれる場合は好まれない。
RNAとタンパク質の両方を含む複合体で、CRISPR-Cas9によるゲノム編集において、RNPは、ハイブリッドを形成したcrRNA、tracrRNA、およびCas9エンドヌクレアーゼから構成される。RNPフォーマットでのCRISPR-Cas9の導入は、ゲノム編集効率を最適化し、オフターゲット効果を低減させる[6]。
CRISPRエンドヌクレアーゼによって特異的な塩基配列。crRNAに含まれ、Cas9エンドヌクレアーゼのPAMは「NGG」である。
crRNAのプロトスペーサー配列(標的配列)と相補的なゲノムDNA鎖。
ターゲット鎖と相補的な、PAM配列を有するゲノムDNA鎖。
一本鎖オリゴヌクレオチド、一本鎖DNA、ssDNA。
二本鎖DNA。
CRISPRを介したゲノム編集実験において、細胞や受精卵に導入するCRISPR試薬等の総称。本文では主にCRISPR-Cas9 NucleaseとcrRNA:tracrRNA の複合体(RNP)を指す。
HDRを最適化するためのcrRNA設計
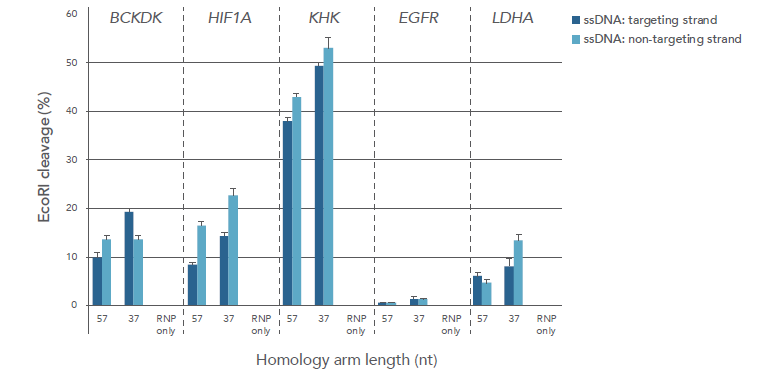
当社では、初回のスクリーニングには、インサートを挿入したい箇所の近傍の、少なくとも2~4か所のCRISPR-Cas9 PAMサイトに対して試験することを推奨しております。図3は、5箇所の異なるPAMサイトにおいて、EcoRI制限酵素の認識部位を含む短いインサートのHDR導入効率を示しております。
HDRによる新規の制限酵素サイトの導入は、RFLP(制限酵素断片長多型)を生じさせ、これにより、制限酵素を用いた切断によるHDR導入の成否の判断が可能となります。EcoRIによるの切断から測定されるHDR効率は、たとえ全ての切断サイトがDSBを効率的に生じても、部位特異的であることは明確です。
現状最も効率の良いCRISPR-Cas9の系であっても、各PAMサイトに対して、必ずしも切断できるとは限りません。しかし、次節で詳細に考察しますが、HDR効率に関しては、HDRテンプレートを合理的に設計することによっておそらく改善することができます。
インサートの導入サイトからgRNAの位置を考慮することも必要です。Cas9タンパク質によって生じるDSBサイトが、インサートの導入予定位置に近接していることが理想的です[7]。
HDR効率は、ドナーテンプレートが切断部位から5~10塩基離れると劇的に低下します(図4)。しかしながら、必ずしも近接のPAMサイトが利用可能とは限りません。このような状況下では、効率的にHDRが生じるように、できるだけ近く、かつ活性の高いPAMサイトを選択してください。
本報で紹介するデータはすべて、CRISPR/Cas9を用いたHDRにおいて有用な、不死化細胞株であるHEK-293に関する研究から得ております。
A. HDR効率は、ドナーテンプレートによって変化する
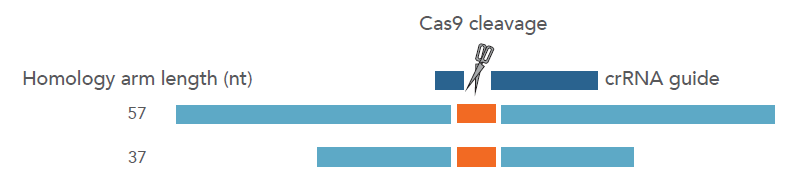
B. HDRテンプレートデザインとCas9の切断サイト
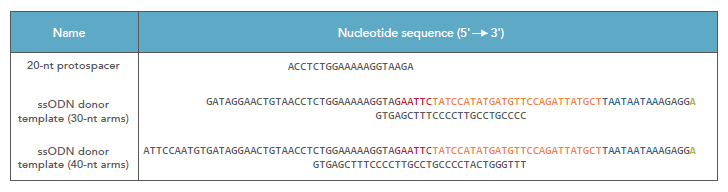
図3. HDR効率はPAMサイトにより変化する。
(A)1.2 µLのLipofectamine® RNAiMAXトランスフェクション試薬(Thermo Fisher)を用いて、HEK-293細胞に、10 nMのRNP(Alt-R CRISPR-Cas9 crRNA:tracrRNA と複合体を形成したAlt-R S.p. Cas9ヌクレアーゼ3NLS)および3 nMのHDRテンプレート(脱塩グレードのUltramerオリゴ)を導入した。リポフェクションの48時間後にゲノムDNAを単離した後、ターゲット領域をPCRで増幅した。PCR産物をEcoRIで切断し、Fragment Analyzer™システム(Advanced Analytical)を用いて分析し、HDR効率としてEcoRI配列の挿入率を求めた。
(B)37 ntまたは57 ntのいずれかの3'および5'ホモロジーアームをEcoRIサイトに隣接するように設計し、ホモロジーアームの長さがどのようにHDRの導入効率に影響するかを調べた。オレンジは、HDR テンプレートにおけるEcoRIサイトを表す。
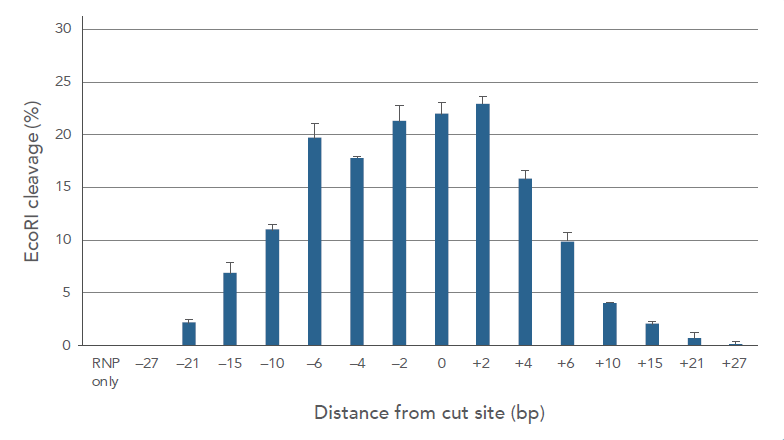
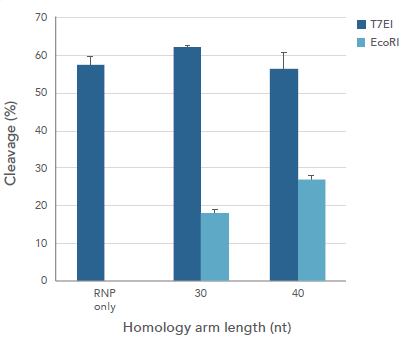
図4. HDRの導入率はCRISPR-Cas9による切断サイトから10塩基以上離れた位置で顕著に低下する
ヒトSERPINC1遺伝子をターゲットとするAlt-R CRISPR-Cas9 RNPを、4 µMの濃度でHEK-293細胞にエレクトロポレーションで導入した。EcoRIサイトとCas9による切断位置とを、図のように様々に配置したドナーテンプレートも同時に導入した。実験に用いたssODNのドナーテンプレートの設計には、40 ntの非ターゲット鎖の配列を用いた。ゲノム編集効率をさらによくするために、2 µMのAlt-R Cas9 エレクトロポレーターエンハンサー(IDT)も加えた。エレクトロポレーションの48時間後にゲノムDNAを単離し、HDR導入率をEcoRIを用いて評価した。
DNAテンプレート:dsDNAとssDNAのテンプレート
さらにdsDNAテンプレートは、培養細胞に対して有害である可能性があります。リニアなdsDNAまたはプラスミドdsDNAの導入では、リポフェクションの効率が低くなったり、細胞毒性が見られることがしばしばあります。
大まかに言うと20 bp未満の塩基置換や小さな導入配列(例. 終止コドン、タンパク質機能部位、エピトープタグ、タグ配列の付加)については、IDTでは、UltramerオリゴヌクレオチドなどのssODNテンプレートを用いることを推奨しております。200 塩基以上についても、最長2000塩基の提供が可能な、Megamer を利用可能です。
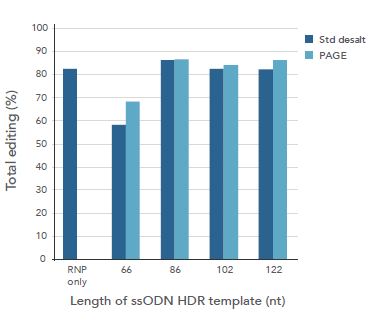 A. 切断効率 — 脱塩グレード vs PAGEグレード
A. 切断効率 — 脱塩グレード vs PAGEグレードssODNテンプレート
B. HDR効率 — 脱塩グレード vs PAGEグレードssODNテンプレート
図5. 脱塩グレードのUltramerは点突然変異や短い配列を導入するための理想的なssODNテンプレートである
10 nMのRNP(Alt-R CRISPR-Cas9 crRNA:tracrRNAと複合体形成したAlt-R S.p. Cas9ヌクレアーゼ3NLS)と、脱塩グレードまたはPAGEグレードのいずれかによって精製した3 nMのUltramerオリゴヌクレオチド(すなわち、ssODN HDRテンプレート)を1.2 µLのCRISPRMAX®(Thermo Fisher)を用いて
HEK-293細胞へ、トランスフェクトした。※66-/102-nt ssODNドナーが30 ntのホモロジーアームを有しているのに対し、86-/122-ntドナーのホモロジーアームは40 ntである。
ゲノム編集を評価するため、ゲノムDNAはトランスフェクション後48時間後に抽出し、ターゲット領域をPCRにて増幅した。その後、NGSを用いてPCR産物を分析した。
アンプリコンをMiSeq®システム(Illumina)でシーケンスした。社内で開発したプロセシングプログラムにより配列決定データを分析して、(A)全体的な切断効率と(B)HDR効率を同時に定量化した。
Std=標準。
ssODNドナーテンプレートのデザイン
当社では今なお、可能な場合は両方の鎖に対してドナーテンプレートを合成することを推奨しております。しかしながら、各PAMサイトに対して導入するドナーテンプレート数が限られている場合、非ターゲット鎖と同一のssODNドナーオリゴを設計することによって、成功率が改良される場合があります。
なお、当社の実験の多くが「短い配列」を挿入していることに留意してください。「長い配列」の挿入については、現在も研究中であり、異なる実験結果を示す場合があります。
図6の通り、Cas9-DSBサイトの中心に、6塩基のEcoRI制限酵素サイトと、それぞれ27~92塩基のホモロジーアームを対象的に配置したHDRドナーテンプレートを6種類設計しました。すでに報告されている通り[11]、ホモロジーアームが30 ~ 60塩基である場合、比較的高いHDR効率が観察されました。
一方、dsDNAドナーテンプレートのHDR効率が高いと判定されたほとんどの場合、導入配列長を確認すると、dsDNAテンプレートがNHEJとHDRの両方の経路で挿入されている事が明らかになりました。 NHEJ経路によって変異を導入しようとした場合には、ssODNをドナーテンプレートとして用いた場合には観察されない、ホモロジーアームの重複が生じます(データ非開示)。
IDTでは、ホモロジーアームを非対称に設計した実験も行いましたが、HDR効率に対する相関は見られませんでした(データ非開示)。
しかしながらこの事象は、一部の生物やタンパク質のアクセスが難しいゲノム領域に対しては有益かもしれませんが、実験がさらに複雑になります。そのため、現在のところIDTでは、HDRの導入においてはこの戦略の採用を控えています。
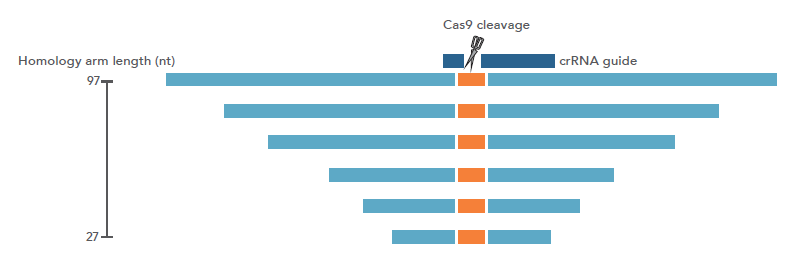
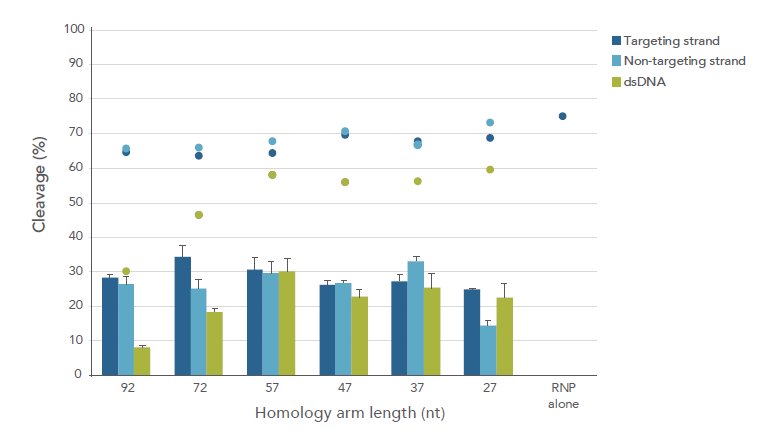
B. HDRドナーテンプレートのデザインとHDR効率
図6. ssODNのホモロジーアームが30~60塩基長のときに、HDR効率が最適化される
(A)EcoRI配列(6塩基)と、両側に27~92塩基長のホモロジーアームを持つHDRドナーテンプレートを6種類設計した。それらのHDRドナーテンプレートはssDNAおよびdsDNAで合成した。
(B)PCR産物をT7EIで切断することで、ゲノム編集効率自体を測定(各ドット)し、EcoRIで制限酵素処理した後のHDR効率も測定した。
ケーススタディ:HDRドナーテンプレートの設計方法
HAタグと3個の終止コドンをHEK293細胞のヒト
図7-Aに示すように、変異の導入部位付近に3箇所のCRISPR-Cas9のPAMサイト(PAM1~PAM3、オレンジ)が利用できるため、これら3サイトをPAMとして選択した。挿入したい42塩基を中心に、30塩基または40塩基の2種のホモロジーアームをもつHDRドナーテンプレートを設計した。(図7-B)。
図7-Cは、2種のssODNを用いた場合のゲノム編集自体の効率とHDRの導入効率を示す。
今回のドナーテンプレートにはPAM配列側の非ターゲット鎖図7-Aを用いたが、ターゲット鎖も用いる事も出来ることを示している。
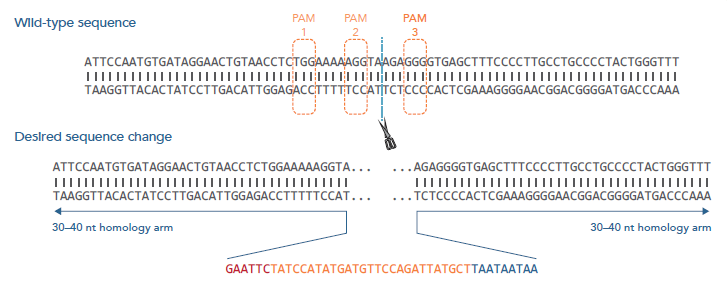
C. ホモロジーアーム長によるHDR効率
図7. HAタグ導入用HDRドナーテンプレートのデザイン
(A,B)インサートを機能によって色分けしている。HDRの検出に使用した6塩基のEcoRI認識部位を赤色、27 塩基のHAタグをオレンジ、3つのTAA終止コドンを青色で示す。
(B)サイレントG→A変異(緑色)をPAMサイトに導入し、DSBが一度修復されたら、切断のターゲットに再度なることを防ぐ。
(C)10 nMのRNP(Alt-R CRISPR-Cas9 crRNA:tracrRNAと複合体形成したAlt-R S.p. Cas9ヌクレアーゼ3NLS)を 、脱塩グレードの3 nMのUltramer HDRドナーテンプレートとともに、HEK-293細胞に、1.2 µLのCRISPRMAX Cas9 Transfection Reagent(Thermo Fisher)を用いて導入した。導入してから48時間後にゲノムDNAを単離し、次いでターゲット領域をPCRで増幅した。PCR産物をT7EIで切断してゲノム編集効率自体(ネイビー)を求め、EcoRI制限酵素処理によりHDR効率(青色)を求めた。
HDRドナーテンプレートデザインのコツ
- 一本鎖のHDRドナーテンプレートは、dsDNAよりも良好な結果が得られる。UltramerをHDRドナーテンプレートとして用いる場合、脱塩グレードを推奨する。ultramerの精製に費用を掛けてもHDRの結果には影響しない。
- できるだけ変異導入部の近くをPAMサイトとして選択する。HDR効率は、切断サイトが変異導入サイトから10塩基以上離れている場合には、有意に低下してしまう。
- HDRドナーテンプレートは挿入部を中心に配置し、その両端に30~60塩基のホモロジーアームをデザインする。小さな配列の挿入や変異であれば、30~60塩基のホモロジーアームで、高いHDR効率が得られる。
- 可能であれば、ssODNでターゲット鎖と非ターゲット鎖の両方に対してHDRドナーテンプレートをデザインする。非ターゲット鎖側のドナーテンプレートの使用は、比較的高いHDR効率が得られるが、必ずしもそうであるとは限らない。
- 一度修復された配列が再び切断されないように、修復した配列をcrRNAによって認識させないようにすることが重要である。認識配列やPAMサイトに対して、1つ以上のサイレント変異を持つHDRドナーテンプレートをデザインすることで、修復後のゲノムの再切断の防止につながる。
導入
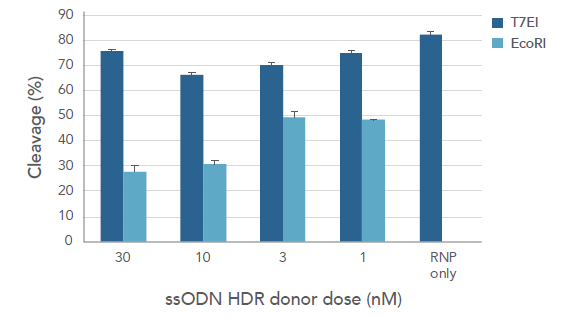
図8-Aは、リポフェクションでRNPを導入した際のHDR導入効率を示しています。
IDTは、HEK-293細胞に対してはAmaxa®のヌクレオフェクション(Lonza)、Jurkat T細胞に対しては、Neon® を用いたエレクトロポレーション(Thermo Fisher)の最適化したプロトコールを開示しています。これらのプロトコールは、お客様のHDR実験の基礎として用いることもできます。
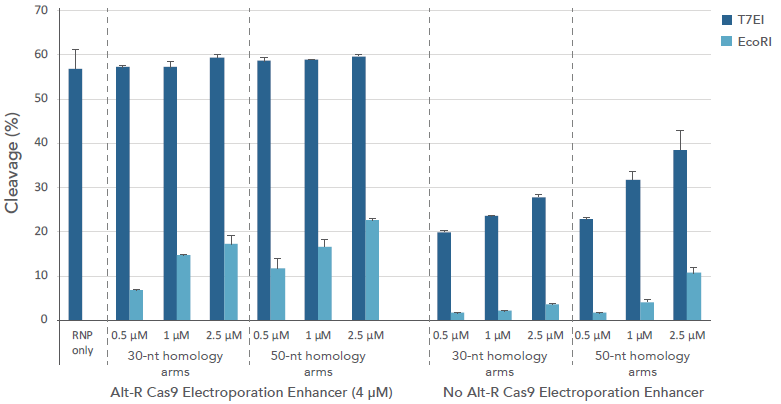
図8-Bに示すように、エレクトロポレーションでも効率よくHDR導入を行えます。なお、エレクトロポレーションの効率を改善するためにデザインされた精製されたキャリアDNAであるAlt-R Cas9 Electroporation Enhancerを用いると優位にHDR効率が向上します。哺乳類の培養細胞以外のマウス、線虫、およびゼブラフィッシュなどの主要なモデル生物において、CRISPRコンポーネントの導入には、マイクロインジェクションも確立されたアプローチです。
B. エレクトロポレーション
図8. リポフェクションまたはエレクトロポレーションを用いた培養細胞に対する効率の良いHDR導入
(A)ヒト
(B)ヒト
ゲノムDNAは導入の48時間後に単離した。ゲノム編集効率自体の効率およびHDR効率をそれぞれ、T7EIおよびEcoRI処理によって評価した。
HDRドナーテンプレート導入ガイドライン
リポフェクションを行う場合、RNPを調製した後、一本鎖HDRテンプレートを1~3 nMの濃度で添加してから、リポフェクション試薬と組み合わせて培養細胞へ添加します。推奨されるリポフェクション実験のセットアップおよびプロトコルについては、Alt-R CRISPR-Cas9 Uder Guide — Alt-R CRISPR-Cas9 system—RNP transfections (recommended)をご参照下さい。
エレクトロポレーションによるCRISPRコンポーネントの導入方法は、機器によって異なります。IDTでは、Amaxa Nucleofector System(Lonza)およびNeon Transfection System(Thermo Fisher)の両方に利用できるプロトコールや、NepaGene、BEX社のエレクトロポレーターのUser-submitted methodsも公開しています。なお、RNP複合体は、ドナーテンプレートの添加前に形成してください。
CRISPR-Cas9系でHDR導入を行う場合、ssODN HDRテンプレートを終濃度2~4 µM、RNPおよびAlt-R Cas9 Electroporation Enhancerを導入用の溶液に添加します。実験のセットアップおよびプロトコールについては、Alt-R CRISPR-Cas9 system—RNP electroporation,Neon Transfection systemや、Alt-R CRISPR-Cpf1—RNP electroporation, Amaxa Nucleofector system を参照してください。
Alt-R CRISPR RNP複合体のマウス受精卵へのマイクロインジェクションは、sgRNAとCas9 mRNAの導入などの他の方法と比較して、おそらくRNPは導入後すぐに切断を行うため、高いHDR効率が得られます。
IDTは複数の共同研究先と、201 塩基以上のssODN(Megamer)をHDRドナーテンプレートとして用いて、他のCRSPRコンポーネントと同時に導入するEasi-CRISPRというマウスのゲノム編集戦略を開発し、ノックインにおけるげっ歯類のモデルの作出しました[10]。HDRにはssDNAは5~20ng/µLを使用して下さい。
マウス受精卵におけるマイクロインジェクションのプロトコールは、CRISPR-Cas9 RNP delivery—mouse zygote microinjection—CB Gurumurthy をご参照下さい。
User Guideおよびプロトコルはすべて、www.idtdna.com/protocolsで公開しています。
HDRによる挿入の確認
ゲノム編集自体の効率を確認することは、HDR効率の指標の一つとなります。 HDR効率の定量化については、以下の方法のうちの1つを採用することができます。
- HDRドナーテンプレートに対して特異的なPCRアッセイをデザインする
プライマーは非特異断片を増幅しないよう、慎重に設計して下さい。例えば、プライマーの1つをHDRドナーテンプレートの外側に、もう1つを挿入する配列内もしくは、挿入配列を含むようにデザインします。 - 制限酵素配列を挿入配列に付加する
制限酵素配列の挿入によって、PCRと制限酵素処理によってHDRを検出できます。この手法は、本研究で弊社が用いた検出アプローチです。 - タグ配列を付加して、フローサイトメトリーなどで検出する
- 編集した領域を増幅するPCRアッセイを設計し、シーケンスを読む
結論
このアプリケーションノートでは、HDR実験の最適化について、当社が数々の実験で得た結果を要約しております。弊社や他の科学コミュニティの方々もHDRについて、特に長鎖の挿入や置換など、まだまだ理解すべきことは数多くあります。
CRISPR研究および開発を続ける限り、このアプリケーションノートを更新し続けます。
要約
- HDR効率のベースラインはエラー率の高いNHEJ経路と比較して低いので、HDR導入実験の条件を最適化することは重要です。
- UltramerやMegamerなどの純度の高いssODNは、HDR実験に有用なドナーテンプレートです。dsDNAドナーテンプレートは、HDR効率が低く、平滑断端としての挿入、および細胞毒性を示します。
- HDR効率は隣接するPAMサイト毎に異なるため、可能であれば複数のPAMサイトで実験を行って下さい。変異導入部位からできるだけ近く、切断活性の高いcrRNAを選択して下さい。
- ssODNテンプレートは、HDR効率が最大化するようにデザインして下さい。
- CRISPRコンポーネントの導入は、対象の生物に対して徹底的な条件検討が必要な場合があります。Alt-R Cas9 Electroporation Enhancerを用いるエレクトロポレーションは、リポフェクションに適していない培養細胞に対する代替的な導入アプローチとして役立ちます。
- 目的の変異の挿入確認の戦略をできるだけ早く考慮してください。実験計画段階での想定が理想てきです。
References
著者:Michael Collingwood, Ashley Jacobi, Bernice Thommandru, Mollie Schubert, Garrett Rettig,
Hans Packer, Ellen Prediger, and Brian Wang
翻訳:安井 孝彰
- Hsu PD, Lander ES, et al. (2014) Development and applications of CRISPR-Cas9 for genome engineering. Cell, 157(6):1262–1278.
- Agudelo D, Duringer A, et al. (2017) Marker-free coselection for CRISPR-driven genome editing in human cells. Nat Methods, 14(6):615–620.
- Lieber MR (2010) The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem, 79:181–211.
- Mao Z, Bozzella M, et al. (2008) Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair (Amst), 7(10):1765–1771.
- Rivera-Torres N, Banas K, et al. (2017) Insertional mutagenesis by CRISPR/Cas9 ribonucleoprotein gene editing in cells targeted for point mutation repair directed by short single-stranded DNA oligonucleotides. PLoS One, 12(1):e0169350.
- Jacobi AM, Rettig GR, et al. (2017) Simplified CRISPR tools for efficient genome editing and streamlined protocols for their delivery into mammalian cells and mouse zygotes. Methods, 121-122:16–28.
- Inui M, Miyado M, et al. (2014) Rapid generation of mouse models with defined point mutations by the CRISPR/Cas9 system. Sci Rep, 4:5396.
- Miura H, Gurumurthy CB, et al. (2015) CRISPR/Cas9-based generation of knockdown mice by intronic insertion of artificial microRNA using longer single-stranded DNA. Sci Rep, 5:12799.
- Yoshimi K, Kunihiro Y, et al. (2016) ssODN-mediated knock-in with CRISPR-Cas for large genomic regions in zygotes. Nat Commun, 7:10431.
- Quadros RM, Miura H, et al. (2017) Easi-CRISPR: A robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins. Genome Biol, 18(1):92.
- Wefers B, Bashir S, et al. (2017) Gene editing in mouse zygotes using the CRISPR/Cas9 system. Methods, 121-122:55–67.
- Renaud JB, Boix C, et al. (2016) Improved genome editing efficiency and flexibility using modified oligonucleotides with TALEN and CRISPR-Cas9 nucleases. Cell Rep, 14(9):2263–2272.
- Zhou J, Wang J, et al. (2014) Dual sgRNAs facilitate CRISPR/Cas9-mediated mouse genome targeting. FEBS J, 281(7):1717–1725.
製品フォーカス
Alt-R™ CRISPR-Cas9 System
Alt-R® CRISPR-Cas9 Systemには、ゲノム編集を行うためのキーとなる試薬が揃っています。S. pyogenesの持つCRISPR-Cas9 systemに由来する本システムは、下記の様に多数のアドバンテージがあります。- 他社の手法と比較し、オンターゲット率が向上します。
- リポフェクションやエレクトロポレーションで、効率よくRNPが導入できます。
- sgRNAやCas9 mRNAを導入した際に見られる、自然免疫を惹起しません。
Alt-R® CRISPR-Cpf1 System
Alt-R™ CRISPR-Cpf1 Systemを用いると、これまでCRISPR-Cas9で切断できなかったサイトでゲノム編集を行えます。Cpf1でDNAを切断すると、5'突出末端となります。本試薬には下記の特徴があります。
- AT-richなゲノム領域でもゲノム編集が行えます。
- Cas9では切断できなかったサイトを補填できます。
- Cpf1 NucleaseとcrRNAのみで、ゲノム編集を行えます。tracrRNAは必要ありません。
- エレクトロポレーション法で効率よくRNPを導入できます。