Q & A サービス内容のご質問
DNA
RNA
Ultramer
PrimeTime
Genes
gBlocks
溶解後は、小分け分注し[-20℃/遮光]にて保管してください。
Q. 保管について
A. 溶解前は[室温保存]で良いですが、長期保存(1ヶ月以上)の場合は[-20℃]で保存下さい。溶解後は、小分け分注し[-20℃/遮光]にて保管してください。
DNA
RNA
Ultramer
PrimeTime
Q. MSAAのデータがきれいですが、純度が良いということですか?
A. MASSのデータは合成されたオリゴの分子量を測定しています。メインのピーク以外はカットオフしていますので、実際の純度を反映するものではございません。純度はCEデータで確認することができます。
DNA
RNA
Ultramer
PrimeTime
Genes
gBlocks
Q. プロトコルにはIDTE Buffer(10mM Tris,0.1mM EDTA,pH8.0)に溶解するとありますが、1mM EDTAで調製されたTE BufferやDWで溶かすことはできませんか。
A. 一般的な濃度のTE (10mM Tris, 1mM EDTA, pH 8.0) もご利用いただけます。IDTではEDTA濃度は0.1mMの薄い濃度でも問題ないこと、キレート剤はPCR反応とトレードオフの関係にあることからこの濃度を採用しています。またDWも構いませんが、TEの方が保存安定性がありますので、TEに溶解することをお薦めしております。
DNA
この処理で溶解する場合には、その後、問題なくご使用いただけます。
Q. 合成DNAが溶解しませんでした。
A. オリゴの配列や修飾により、極稀に起こる現象です。溶解しにくい場合は、「65度で20分間インキュベート」で解決できる可能性がありますのでお試し下さい。この処理で溶解する場合には、その後、問題なくご使用いただけます。
PrimeTime
3-10 min, 95℃
15 sec, 95℃ ⇒ 45 sec, 60℃ 40サイクル
Q. IDT推奨のPCRサイクルを教えてください。
A. 下記を標準条件として検定しています。3-10 min, 95℃
15 sec, 95℃ ⇒ 45 sec, 60℃ 40サイクル
DNA
PrimeTime
プライマー:62 ±2℃ (60℃~64℃)
プローブ:68 ±2℃ (66℃~70℃)
Q. IDT推奨のTm値はありますか?
A. 下記が理想です。プライマー:62 ±2℃ (60℃~64℃)
プローブ:68 ±2℃ (66℃~70℃)
DNA
PrimeTime
https://sg.idtdna.com/calc/analyzer
このページのパラメーターを任意にセットし、Analyzeボタンを押しますと、Tm値計算結果が表示されます。
なお、定量PCR用のプライマーおよびプローブのTm値を計算する際には、以下のパラメーターを使います。
Oligo concentration 0.2 µM
Na+ concentration 50 mM
Mg++ concentration 3 mM
dNTPs concentration 0.8 mM
Q. どのようにTm値を計算していますか?
A. より高い精度のTm値設定を行えるようにするため、独自のアルゴリズムで計算するTm値計算ツールを用意しています。https://sg.idtdna.com/calc/analyzer
このページのパラメーターを任意にセットし、Analyzeボタンを押しますと、Tm値計算結果が表示されます。
なお、定量PCR用のプライマーおよびプローブのTm値を計算する際には、以下のパラメーターを使います。
Oligo concentration 0.2 µM
Na+ concentration 50 mM
Mg++ concentration 3 mM
dNTPs concentration 0.8 mM
PrimeTime
※文中には、使用する溶解バッファーとして「IDTE buffer」と 記載がありますが、通常のTE bufferもご利用いただけます。
Q. PrimeTime®の溶解方法について
A. 溶解プロトコール(英語)をご参照下さい。※文中には、使用する溶解バッファーとして「IDTE buffer」と 記載がありますが、通常のTE bufferもご利用いただけます。
PrimeTime
http://sg.idtdna.com/pages/docs/default-source/catalog-product-documentation/primetime-pre-designed-qpcr-assays-brochure.pdf?sfvrsn=8
もしうまくワークしない場合、トラブルシューティング(再合成や別のセットの提案)を行いますので、お気軽にお知らせ下さい。トラブルシューティングの際は、コントロール実験の結果をお知らせ下さい。
Q. プレデザインはバリデート済みですか?
A. 全てのプレデザインについてバリデートは行っておりませんが、ランダムに選んだ150セットを試験したところ、99%以上のセットがPCR効率90%以上でワークした、という結果は得ております。http://sg.idtdna.com/pages/docs/default-source/catalog-product-documentation/primetime-pre-designed-qpcr-assays-brochure.pdf?sfvrsn=8
もしうまくワークしない場合、トラブルシューティング(再合成や別のセットの提案)を行いますので、お気軽にお知らせ下さい。トラブルシューティングの際は、コントロール実験の結果をお知らせ下さい。
PrimeTime
Q. IDTでは、推奨プロトコールがありますか?
A. IDTでは、PrimeTime® qPCR assay に下記のプロトコールを推奨しております。(p.44)
PrimeTime
Q. 内在性コントロールについて、推奨はありますか。
A. ヒト・マウス・ラットについては、下記表をご覧ください。| ヒト | マウス | |||||
| Accession番号 | 遺伝子名 | PrimeTime Assay ID | Accession番号 | 遺伝子名 | PrimeTime Assay ID | |
|---|---|---|---|---|---|---|
| NM_000194(1) | HPRT1 | Hs.PT.58v.45621572 | NM_009735(1) | B2m | Mm.PT.58.10497647 | |
| NM_004048(1) | B2M | Hs.PT.58v.18759587 | NM_013684(1) | Tbp | Mm.PT.58.10867035 | |
| NM_003194(1) | TBP | Hs.PT.58v.39858774 | NM_010368(1) | Gusb | Mm.PT.58.13834361 | |
| NM_000181(1) | GUSB | Hs.PT.58v.27737538 | NM_007393(1) | Actb | Mm.PT.58.33257376.gs | |
| NM_001101(1) | ACTB | Hs.PT.56a.40703009.g | NM_009089(1) | Polr2a | Mm.PT.58.13811327 | |
| NM_000937(1) | POLR2A | Hs.PT.58.25515089 | NM_007475(1) | Rplp0 | Mm.PT.58.43894205 | |
| NM_001002(2) | RPLP0 | Hs.PT.58.20222060 | NM_008907(1) | Ppia | Mm.PT.39a.2.gs | |
| NM_021130(1) | PPIA | Hs.PT.58v.38887593.g | ||||
| ラット | |||
| Accession番号 | 遺伝子名 | PrimeTime Assay ID | |
|---|---|---|---|
| XM_003752155(2) | Hprt1 | Rn.PT.58.37395539 | |
| NM_012512(1) | B2m | Rn.PT.58.44337940 | |
| NM_001004198(1) | Tbp | Rn.PT.58.18641244 | |
| NM_017015(1) | Gusb | Rn.PT.58.37252396 | |
| NM_031144(1) | Actb | Rn.PT.58.13971699.gs | |
| XM_002727723(4) | Polr2a | Rn.PT.58.35295130 | |
| NM_022402(1) | Rplp0 | Rn.PT.58.45174577 | |
| NM_017101(1) | Ppia | Rn.PT.39a.22214830 | |
PrimeTime
弊社でもマスターミックスを販売しておりますので、是非ご参照下さい。
>> PrimeTime® Gene Expression Master Mix : プローブ法の定量PCR用マスターミックス
Q. マスターミックスはどちらの社の物を使えばよいですか?
A. 各社のマスターミックスが使用できます。弊社でもマスターミックスを販売しておりますので、是非ご参照下さい。
>> PrimeTime® Gene Expression Master Mix : プローブ法の定量PCR用マスターミックス
PrimeTime
この様にTm値が低いプローブは、正常にワークしないと考えられます。
Minor Groove Binder(以下、MGB) 修飾を施したプローブであれば、正常なTm値(68℃±2℃)を取ることが出来ますが、IDT社では取り扱いがございません。
しかしながら、下記にて同様の結果を得ることが可能です。
1-a) プローブの前後を伸長させ、Tm値を上昇させる方法
Q. 論文から引用した配列のTm値 が極端に低い場合 (MGB修飾プローブの代替方法について)
A. お客様よりご発注頂くプローブの中には、稀にTm値が極端に低い物がございます。(10~15mer 程度)この様にTm値が低いプローブは、正常にワークしないと考えられます。
Minor Groove Binder(以下、MGB) 修飾を施したプローブであれば、正常なTm値(68℃±2℃)を取ることが出来ますが、IDT社では取り扱いがございません。
しかしながら、下記にて同様の結果を得ることが可能です。
- プローブの配列を変更してもよい場合
- a) プローブの前後を伸長させ、Tm値を上昇させる方法
- b) ターゲット遺伝子の異なる箇所で、プライマー・プローブを設計する方法
- プローブの配列が変更できない場合
- a)LNAという修飾塩基を用いる方法
1-a) プローブの前後を伸長させ、Tm値を上昇させる方法
引用論文と同領域にてプローブを作成したい、プローブを伸長させる余裕がある場合は、この方法がお勧めです。
配列を前後に伸長させ、適度なTm値のプローブを設計して下さい。
また、下記より必要事項をお送り頂けましたら、弊社にて設計も可能です。
1-b) ターゲット遺伝子の異なる箇所で、プライマー・プローブを設計する方法配列を前後に伸長させ、適度なTm値のプローブを設計して下さい。
また、下記より必要事項をお送り頂けましたら、弊社にて設計も可能です。
| MGB | |
|---|---|
| Gene NO.MGB atgctcggctctagcatgacaga ctacgtacaatcgaaagctg... |
|
論文と同遺伝子を検出できれば良い場合は、こちらの方法がお勧めです。
下記の必要事項をお送り頂ければ、弊社から配列のデザインをお送り致します。
ヒト・マウス・ラットの遺伝子の場合は、IDTでプレデザインされた物もございます。
2-a) LNAという修飾塩基を用いる方法下記の必要事項をお送り頂ければ、弊社から配列のデザインをお送り致します。
ヒト・マウス・ラットの遺伝子の場合は、IDTでプレデザインされた物もございます。
| Accession NO. | |
|---|---|
| NM_000008 NM_000009 : |
|
SNP解析など、プローブの配列を変更したくない場合は、この方法が有用です。
この修飾を用いることで、引用論文とほぼ同じ配列で、同等のTm値を得る事が可能です。
IDTではLNAプローブにも安価なMiniスケールを設定していますので、お試しで使用したい場合、あるいは数回の実験で十分な場合などにお勧めです。
詳細は、こちらのLNA項をご覧ください。
この修飾を用いることで、引用論文とほぼ同じ配列で、同等のTm値を得る事が可能です。
IDTではLNAプローブにも安価なMiniスケールを設定していますので、お試しで使用したい場合、あるいは数回の実験で十分な場合などにお勧めです。
詳細は、こちらのLNA項をご覧ください。
PrimeTime
PrimeTimeに切り替えの場合は、配列長を長くするかLNAプローブのご使用をお薦めしております。
IDTにツールを用意しておりますので、以下のツールをご活用ください。
● 配列長を長くする場合
http://sg.idtdna.com/calc/analyzer
「Parameter sets」を「qPCR」にしてご使用してください。IDT推奨のプローブのTm値は68 ±2℃ (66℃~70℃)です。
配列長を長くする方をご選択の場合、配列が長くなると蛍光とクエンチャーの距離が離れるため、バックグラウンドの蛍光値が上がる傾向があります。
その場合は、ZENというInternal quencher(IDTオリジナル)を9塩基と10塩基の間に挿入して、バックグラウンドを抑えることをお薦めしております。
ZENとセットで使用できるクエンチャーはIowa Black(IDTオリジナル)です。
ちなみに、ZEN自体はTm値を高めるような効果がありますので、ZENを入れることでハイブリダイゼーションは安定しますが、MGBほどの劇的な効果ではないため、Tm値の計算にZENの効果は考慮しておりません。
● LNA Probesを利用する場合
LNA ProbesのTm値計算ツール
http://biophysics.idtdna.com/
上記サイトの使い方はこちら
http://sg.idtdna.com/jp/site/How_to_design_LNA_probes.html
● デザイン依頼をしたい場合やその他ご質問は、メールにてお問い合わせください。
japan-cc@idtdna.com
Q. MGBプローブからの切り替えを検討しています。そのままの配列で注文できますか?
A. MGBプローブは、Tm Enhancerである特殊な構造(MGB)が3’末端に修飾されているので、そのままの配列ではTm値が低くなってしまいます。PrimeTimeに切り替えの場合は、配列長を長くするかLNAプローブのご使用をお薦めしております。
IDTにツールを用意しておりますので、以下のツールをご活用ください。
● 配列長を長くする場合
http://sg.idtdna.com/calc/analyzer
「Parameter sets」を「qPCR」にしてご使用してください。IDT推奨のプローブのTm値は68 ±2℃ (66℃~70℃)です。
配列長を長くする方をご選択の場合、配列が長くなると蛍光とクエンチャーの距離が離れるため、バックグラウンドの蛍光値が上がる傾向があります。
その場合は、ZENというInternal quencher(IDTオリジナル)を9塩基と10塩基の間に挿入して、バックグラウンドを抑えることをお薦めしております。
ZENとセットで使用できるクエンチャーはIowa Black(IDTオリジナル)です。
ちなみに、ZEN自体はTm値を高めるような効果がありますので、ZENを入れることでハイブリダイゼーションは安定しますが、MGBほどの劇的な効果ではないため、Tm値の計算にZENの効果は考慮しておりません。
● LNA Probesを利用する場合
LNA ProbesのTm値計算ツール
http://biophysics.idtdna.com/
上記サイトの使い方はこちら
http://sg.idtdna.com/jp/site/How_to_design_LNA_probes.html
● デザイン依頼をしたい場合やその他ご質問は、メールにてお問い合わせください。
japan-cc@idtdna.com
PrimeTime
Q. SNPs解析など、ターゲット配列内の1~数塩基の違いの検出方法
A. SNPs解析などの塩基のわずかな違いを検出するには、各アリルにマッチする配列で異なる蛍光色素(通常はFAMとHEX)のプローブ2本を使います。その際LNA塩基など、Tm値を上昇させる修飾を加える事で、より感度をあげる事が可能です。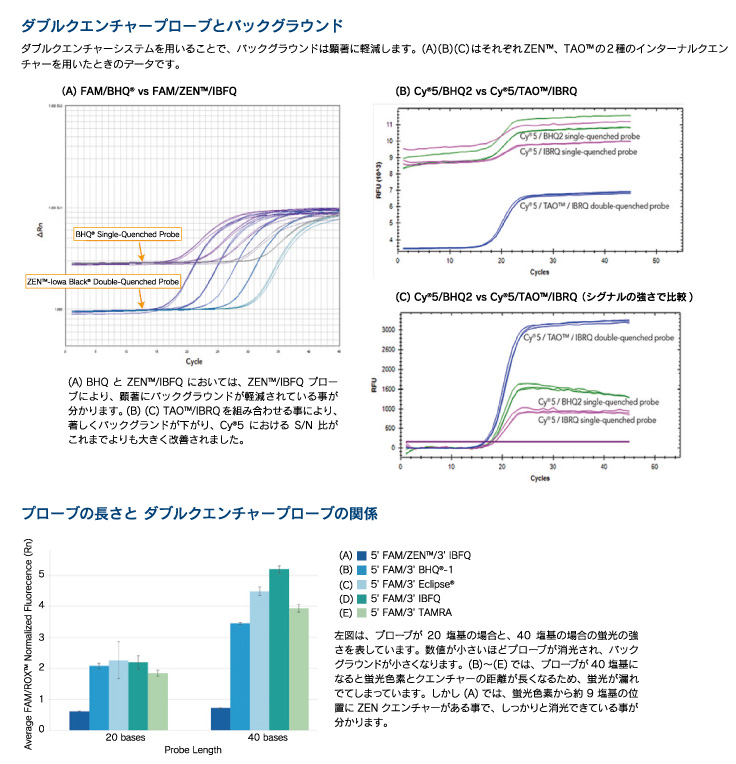
PrimeTime

Q. 色素と対応機器について
A.
PrimeTime
特に、今までクエンチャーとしてTAMRAを用いており、初めてダーククエンチャーを用いるときには、この設定変更についてご注意ください。
Q. ダーククエンチャー(IBFQ,IBRQ,BHQなど)を用いる場合の設定について
A. IBFQなど、蛍光を発しないダーククエンチャーを使う場合、検出機器のクエンチャーの指定を"None"あるいは"Non Fluorescent Quencher"など「蛍光を発しない」という事を意味する設定を指定してください。特に、今までクエンチャーとしてTAMRAを用いており、初めてダーククエンチャーを用いるときには、この設定変更についてご注意ください。
PrimeTime
なお、プライマーとプローブが3本まとめて1本のチューブに入って納品のPrimeTime qPCR Assayを用いる場合は、X10、X20、あるいはX40のマスターストックとして準備し、その倍数に従って反応系を用意してください。
Q. プライマーとプローブの濃度について
A. マスターミックスのプロトコールにプライマーとプローブの濃度の指定が無い場合は、プライマー 500nM、プローブ250nMの濃度でお使いください。なお、プライマーとプローブが3本まとめて1本のチューブに入って納品のPrimeTime qPCR Assayを用いる場合は、X10、X20、あるいはX40のマスターストックとして準備し、その倍数に従って反応系を用意してください。
Genes
ただ、プラスミド骨格部分(ベクター)は転売やキット構成品の販売などの商業利用を制限させて頂いております。
そのため、納品されたプラスミドそのもの (合成断片が、IDT社のベクターに入った状態)では、
research purposes onlyという制限が付きます。
なお、合成した配列部分には使用制限はありません。
上記を明文化した文章はこちら → Vector_Licence (PDFファイル:78KB)
Vector_Licence (PDFファイル:78KB)
Q.ライセンスについて
A. 納品されるプラスミド(合成断片およびベクター)は、研究用途ではライセンスフリーです。ただ、プラスミド骨格部分(ベクター)は転売やキット構成品の販売などの商業利用を制限させて頂いております。
そのため、納品されたプラスミドそのもの (合成断片が、IDT社のベクターに入った状態)では、
research purposes onlyという制限が付きます。
なお、合成した配列部分には使用制限はありません。
上記を明文化した文章はこちら →
Vector_Licence (PDFファイル:78KB)
Genes
クローニングがなかなか成功しない場合にはNew England Biolabsの10 beta株を使用しています。
Q.クローニングに用いる大腸菌について
A. XL1-Blue株を第一選択として用いています。クローニングがなかなか成功しない場合にはNew England Biolabsの10 beta株を使用しています。
Genes
gBlocks
1本鎖DNA/RNAオリゴやgBlocks® Libraries(2本鎖DNAフラグメント)では対応可能です。
Q.「N」や「W」などの混合塩基(mix bases)に関して
A. 申し訳ございませんが、人工遺伝子合成では混合塩基に対応できません。1本鎖DNA/RNAオリゴやgBlocks® Libraries(2本鎖DNAフラグメント)では対応可能です。
Genes
Q.溶解方法について
A.- 溶解前に遠心を行い、DNAペレットをチューブの底に落として下さい。
- 最終濃度が100ng/µLとなるように、TE buffer (10 mM Tris, 0.1 mM EDTA, pH 7.5~8.0)もしくは、nuclease free waterを加えて下さい。
- 30分間の室温静置後、20秒間のボルテックスを行なって下さい。
- 10,000gで1分間遠心後、-20℃で分注及び保管を行なって下さい。
Genes
※希釈倍率が低く、非常に濃いDNA溶液の場合、上手く形質転換出来ないことがあります。
Q.形質転換について
A.- 999µL≒1mLのnuclease free waterを入れたチューブへ、100ng/µLのストックから1µLを移し、100pg/µLのDNA希釈溶液を作ります。
- 1mLのDNA希釈溶液のうち1~2µLを大腸菌への形質転換に用います。
※希釈倍率が低く、非常に濃いDNA溶液の場合、上手く形質転換出来ないことがあります。
Genes
data
なお、波形データの納品は"Genes(人工遺伝子合成)"のみとなります。
gBlocksではデータ納品はございませんので、ご了承下さい。
※ 関連:納品データの取得方法
Q. 納品データに関しまして
A. データは下図の形でお送り致します。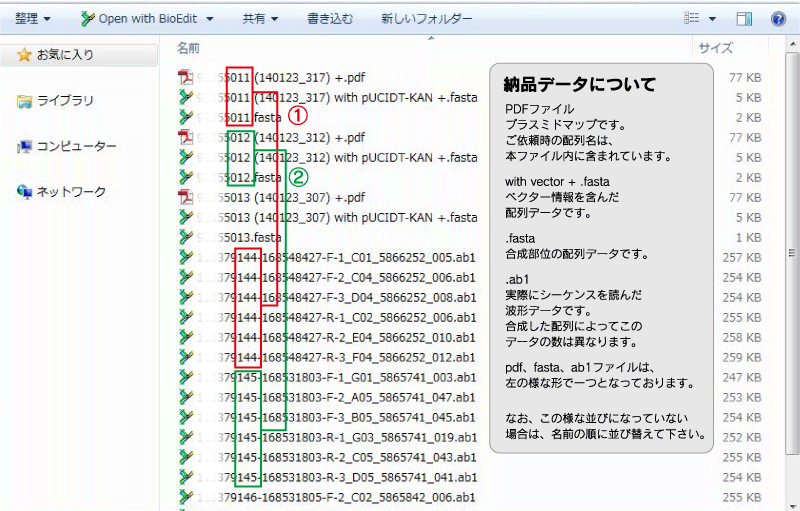
なお、波形データの納品は"Genes(人工遺伝子合成)"のみとなります。
gBlocksではデータ納品はございませんので、ご了承下さい。
※ 関連:納品データの取得方法
gBlocks
また合成できないリスクも上がりますので、例えば2 kbの配列を得たい場合は、1 kbの配列を2本発注し、Gibson Assembly法やIn-Fusion法で繋ぎ合わせるのがお勧めです。
また合成困難性がある場合は、正確な配列の割合が下がりますので、できるだけ排除してください。
Q.gBlocks®の正確性について
gBlocks®は短い方が、合成の正確性は高いです。また合成できないリスクも上がりますので、例えば2 kbの配列を得たい場合は、1 kbの配列を2本発注し、Gibson Assembly法やIn-Fusion法で繋ぎ合わせるのがお勧めです。
また合成困難性がある場合は、正確な配列の割合が下がりますので、できるだけ排除してください。
gBlocks
Q.合成に失敗した配列に共通する特徴はありますか?
長い配列の場合、弱い合成困難性があると合成に失敗するケースがあります。弱い合成困難性であっても出来るだけ除くようにして下さい。
DNA
RNA
Ultramer
PrimeTime
Genes
gBlocks
Q.納品時のオリゴヌクレオチドはどのような状態ですか?
A. 出荷前に溶媒をすべて乾燥させた空気乾燥品です。もし納品物に溶液が残っている場合はすぐにご連絡ください。
DNA
RNA
Ultramer
>> CEデータの取得方法はこちら
Q.精製を行った場合、純度は分かりますか?
A. 15 ~ 60 bases までの合成の場合は、CEデータを取得できますので、こちらで純度をご確認ください。ただし合成が難しい配列や、混合塩基を含む配列など、まれにCEデータを提供できない場合があります。>> CEデータの取得方法はこちら
DNA
ただし、収量が小さく、修飾との相性も良くないため、60 mer以上や、高い純度を必要とする一本鎖オリゴにおすすめです。
Q.HPLC精製とPAGE精製はどのように使い分ければよいですか?
A. HPLC精製は、各種修飾のオリゴに適用可能で、収量も比較的大きいので精製オプションを検討する際の第一選択肢です。>85%の純度が期待できます。PAGE精製は、60 merより長い配列にお勧めです。 >90%の純度が期待できます。ただし、収量が小さく、修飾との相性も良くないため、60 mer以上や、高い純度を必要とする一本鎖オリゴにおすすめです。
DNA
RNA
Ultramer
5'- TTT CGA GTG ATG GTT GTG CCT GCA CGA GAT TCA CAT TCA CGT CTC GGG AA -3'
>> こちらのスペックシートより、さらに詳細なデータをご覧頂けます
Q.収量単位: ODって何ですか?
A.Optical Density: 光学濃度の略称です。ODを測定することで、溶液中のDNA/RNA量を定量することができます。配列ごとにODは異なり、例えば、下記配列を合成した場合は、2.1 nmoles/OD です。5'- TTT CGA GTG ATG GTT GTG CCT GCA CGA GAT TCA CAT TCA CGT CTC GGG AA -3'
>> こちらのスペックシートより、さらに詳細なデータをご覧頂けます
DNA
RNA
Ultramer
Q.TE等のバッファーに溶解して納品することは出来ますか?
A. チューブの場合は1本500円(税別)、プレートの場合は1オーダー20,000円(税別)の追加費用がかかります。チューブの場合は室温での納品、プレートの場合は冷凍品での納品となります。
DNA
RNA
Q.合成スケールってなんですか?
A. 合成開始時に用いる担体のヌクレオチドの量です。実際に納品する量ではありません。
DNA
RNA
Ultramer
dA 313.2
dC 289.2
dG 329.2
dT 304.2
Q.IDTにおける分子量の理論値について
A. IDTでは、DNAのA, T, G, C の分子量には以下の数字を用いています。dA 313.2
dC 289.2
dG 329.2
dT 304.2
※ なお5‘末端の塩基は、リン酸基がついていないため上記の数字よりも62小さくなります。
参考URL:http://sg.idtdna.com/pages/support/technical-vault/reading-room/quick-reference/quick-reference/2011/06/02/molecular-weight-(anhydrous)
参考URL:http://sg.idtdna.com/pages/support/technical-vault/reading-room/quick-reference/quick-reference/2011/06/02/molecular-weight-(anhydrous)
DNA
RNA
Ultramer
Q.混合塩基の分子量に関しまして
A. 例えばNについては A,T,G,C 4つの分子量の平均値(308.95)を用いています。
DNA
RNA
Ultramer
Q.混合塩基におけるGC含率について
A. スペックシートに記載しておりますGC含率に関しまして、例えばNについては、ATが0.5個、GCが0.5として計算しています。
Ultramer
Q.201bases 以上は合成できませんか?
A. 大変申し訳ございませんが、201bases以上は合成できません。
Ultramer
その他のアプリケーションでお使いの場合は、弊社までお問い合わせ下さい。

Q.CRISPR/CAS9に用いる場合、脱塩とPAGE精製どちらが良いですか?
A. 培養細胞に対しては、脱塩でも良いですが、マウス胚へのインジェクションであれば、PAGE精製がお勧めです。その他のアプリケーションでお使いの場合は、弊社までお問い合わせ下さい。